过去,检测这些大小各异的变异需要使用多种不同的检测方法。通过结合因美纳全基因组测序(WGS)与DRAGEN Bio-IT平台内置的二级分析算法,研究人员可以借助混合方法对这些不同类型的所有变异进行高灵敏度检测。DRAGEN v4.2首次引入了一个新选项,支持将基于覆盖度的拷贝数变异(CNV)检出和断裂端结构变异(SV)结果相结合,可更好地解析中小型基因组的增加和丢失事件。这类变异带来了巨大的技术挑战,因为它介于传统测序方法和芯片方法(分别用于较小和较大片段变异)之间。
从数学角度来看,覆盖深度在检测较小事件时的准确性会受到随机波动的干扰,信号变得更加杂乱(如图1所示)。对于大于100 kb的大型事件,这种噪音基本可以忽略不计。在10-100 kb的范围内,尽管存在噪音,但通常不会造成问题。而在1-10 kb的范围内,噪音非常高,出现假阴性和假阳性结果的风险也显著增加。
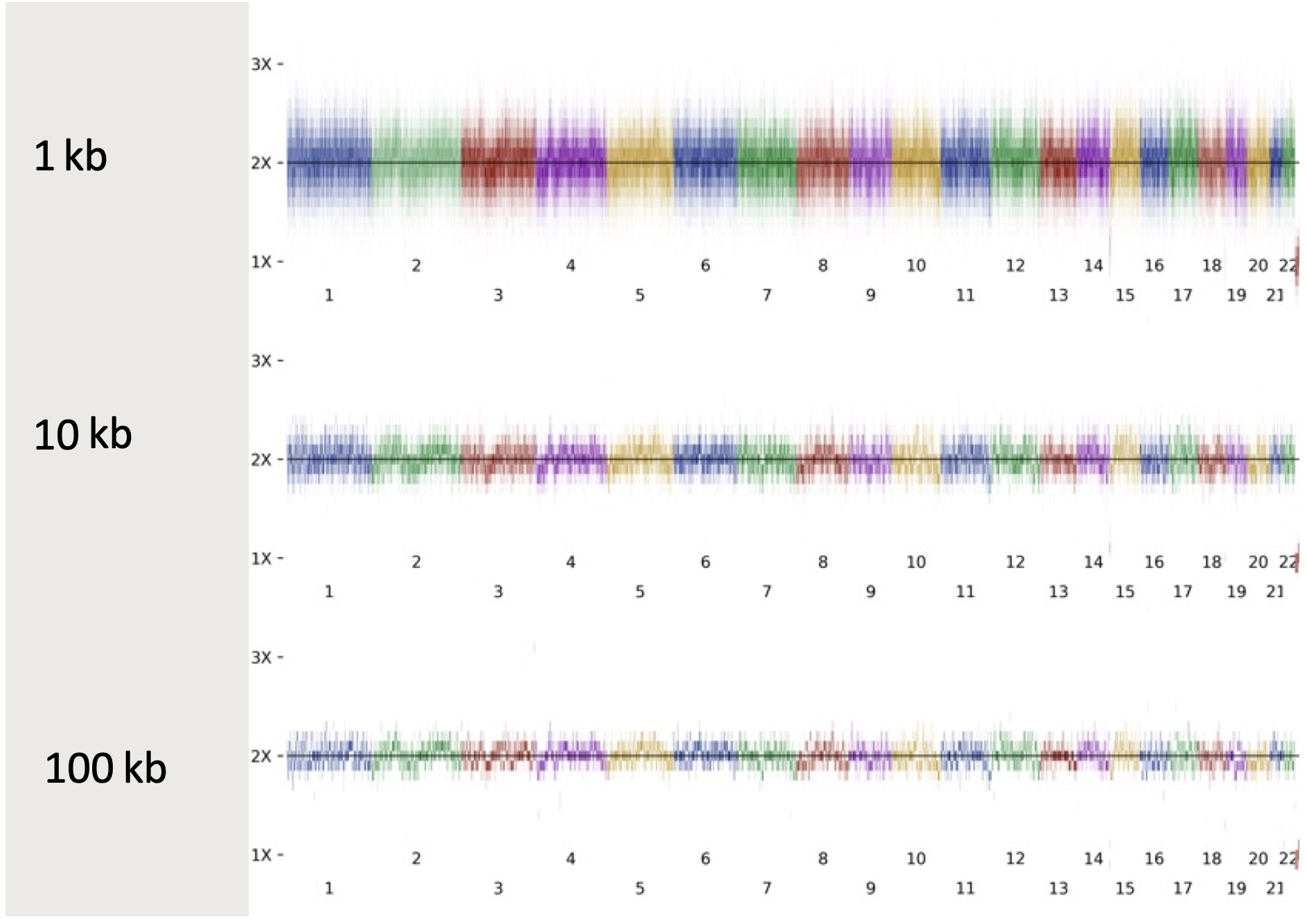
为了解决这一噪音问题,DRAGEN v4.2联合分析了来自生殖系CNV和SV检出程序的信号,识别假定的匹配,更新注释、过滤程序、评分并输出优化的记录。该方法利用来自SV检出程序的连接信号和来自CNV检出程序的深度信号,可以实现低至1 kb CNV的灵敏检测,同时还提高了所有片段长度的查全率和查准率。这是通过结合多重信号证据来改善先前质量低的检测结果,并将CNV断裂端调整为更准确的SV断裂端来实现的。
WGS和DRAGEN整合了几项关键功能,可支持拷贝数和结构变异的高性能分析:
- 使用PCR-free文库制备方法实现整个基因组的均匀覆盖深度,可实现1 kb区间大小下的无对照归一化
- 覆盖内含子和基因间区域的序列数据支持直接观察断点read
- 使用大型数据集进行算法优化,实现严格的过滤调整
- 基于覆盖度和断点的检出程序之间的协同作用可以提高质量评分并完善事件终点
在2023年11月分子病理学大会上,Francisco De La Vega博士(来自Tempus实验室)、Sean Irvine博士(来自Real Time Genomics)和因美纳的Sean Truong共同展示了一项研究,该研究挑战了在医学相关基因中检测中小型CNV的工作。研究结果显示,因美纳全基因组测序和DRAGEN技术相结合,可以成功识别所有引入的变异(图2)。
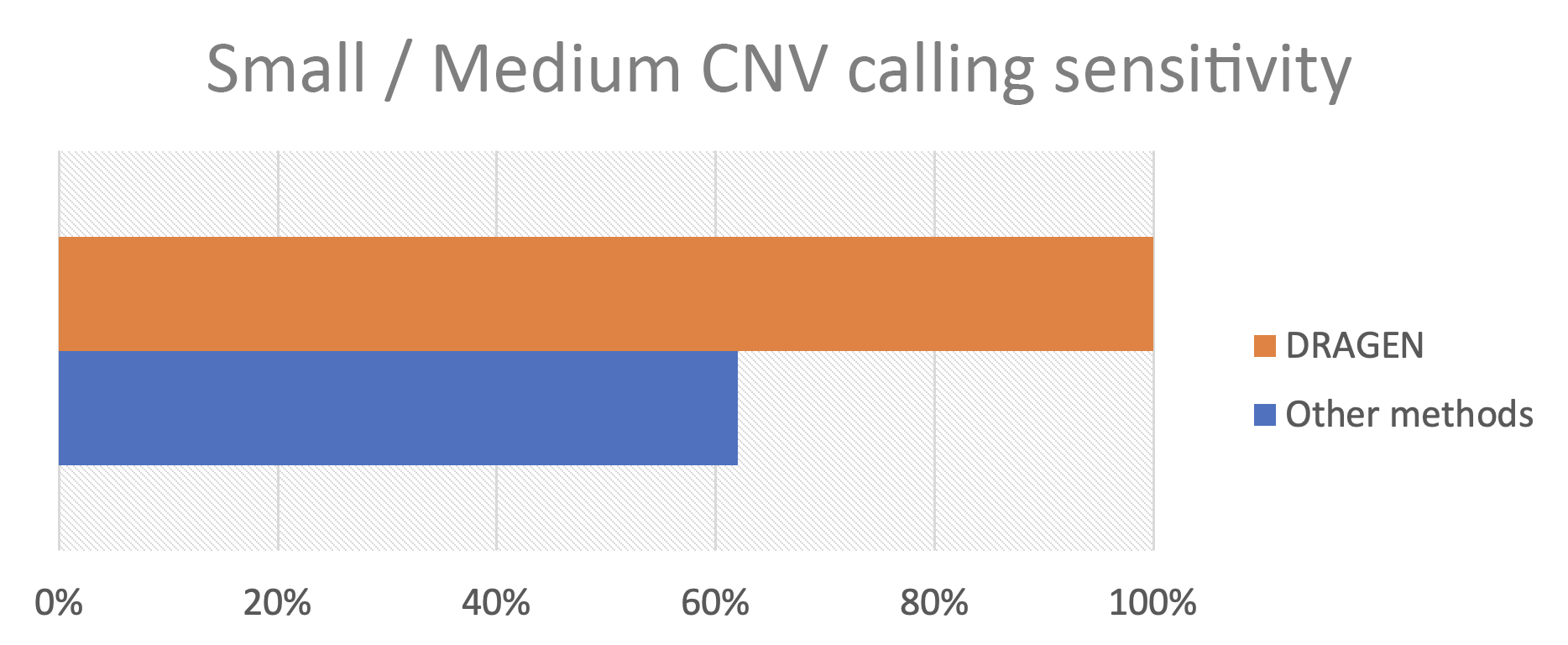
Broad研究所的研究人员还发现,DRAGEN 4.2能够对此大小范围内的事件进行准确的CNV检出,特别是对于5-10 kb之间的缺失事件。
这些结果清晰表明:基因组测序是检测中小型CNV的高灵敏度平台,这类变异对于癌症体质倾向、心血管疾病、携带者生殖筛查等研究和医学应用至关重要。这是DRAGEN科学家和合作伙伴在改进基于覆盖度的变异检出和断裂端变异检出方面不懈努力多年后取得的全新高度。我们对因美纳社区使用这项更新技术可能带来的应用前景充满期待。
请关注我们后续的文章,我们将为您更详细地介绍CNV和SV检出的其他应用。