Introduction
Cell-free DNA (cfDNA) circulating in the blood can arise from various tissues, tumors, or microorganisms present in the body. Liquid biopsy analysis of cfDNA in blood is a relatively noninvasive method for assessing and monitoring certain diseases, including cancer. 1 Tumors shed DNA, referred to as circulating tumor DNA (ctDNA), but it represents a small fraction of the total cfDNA present in blood. Therefore, a robust assay is needed to detect variants in the low levels of ctDNA in the bloodstream.
Next-generation sequencing (NGS) methods enable highly sensitive and specific detection and sequencing of variants in ctDNA. A major challenge with sequencing ctDNA stems from its rarity. The ability to identify low frequency variants from low amounts of ctDNA is confounded by various errors and artifacts introduced during library prep, PCR amplification, and sequencing, making it difficult to distinguish true signal (genetic variants) from all of the noise. In this article, we present a method using unique molecular identifiers (UMIs) and the power of the DRAGEN platform to reduce noise and errors to enable accurate somatic variant calling in ctDNA.
Methods
Nucleic acid preparation
cfDNA was extracted from 2.0-6.0 ml of plasma collected in Streck or EDTA blood tubes using the QIAamp Circulating Nucleic Acid Kit (QIAGEN, Catalog no. 55114). The extracted cfDNA was quantified by capillary electrophoresis using the Fragment Analyzer System (Agilent Technologies, Inc.) targeting the region between 75-250 bp.
Library preparation
Sequencing libraries were generated using the TruSight™ Oncology 500 ctDNA Kit (48 samples, Catalog no. 20039252) with 30 ng input of cfDNA material per sample. DNA fragments were end-repaired, A-tailed, and ligated to adapters before sample barcoding via PCR. Adapters containing unique molecular identifiers (UMIs) were ligated to DNA fragments and duplex barcodes were added during PCR amplification for error correction. Enrichment of targeted regions for all samples required two hybridization steps at 57°C using TruSight Oncology index PCR products. The AccuClear Ultra High Sensitivity dsDNA Quantitation Kit (Biotium, Catalog no. 31028-T) was used to ensure sufficient yield of the post-enriched libraries. Post-enrichment libraries were normalized using bead-based normalization, pooled in equal volumes, denatured, and diluted to the appropriate loading concentration.
Sequencing
Prepared libraries were sequenced with 151 bp paired-end reads on the NovaSeq 6000 System using the NovaSeq 6000 S4 Reagent Kit v1.5 (300 cycles, Catalog no. 20028312) following the XP workflow for individual lane loading (6-plex per lane, 24-plex per flow cell). On average, each sample yielded ~1B reads per library.
Data analysis
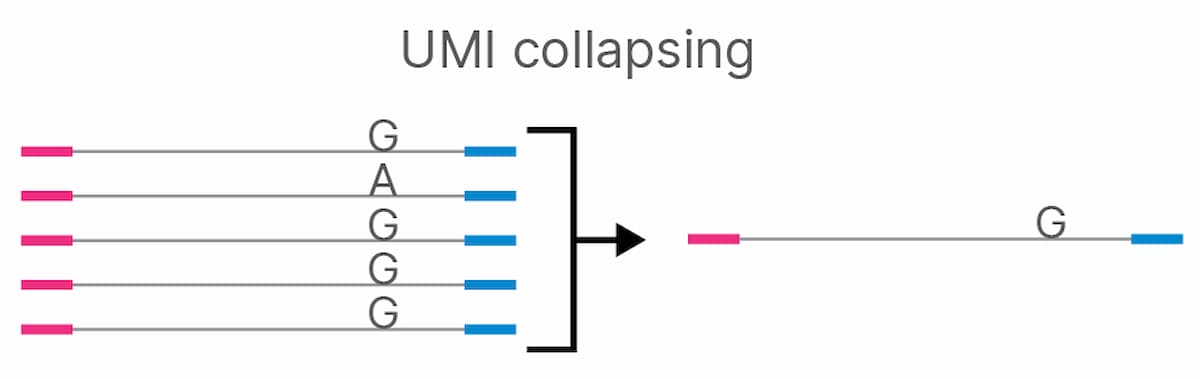
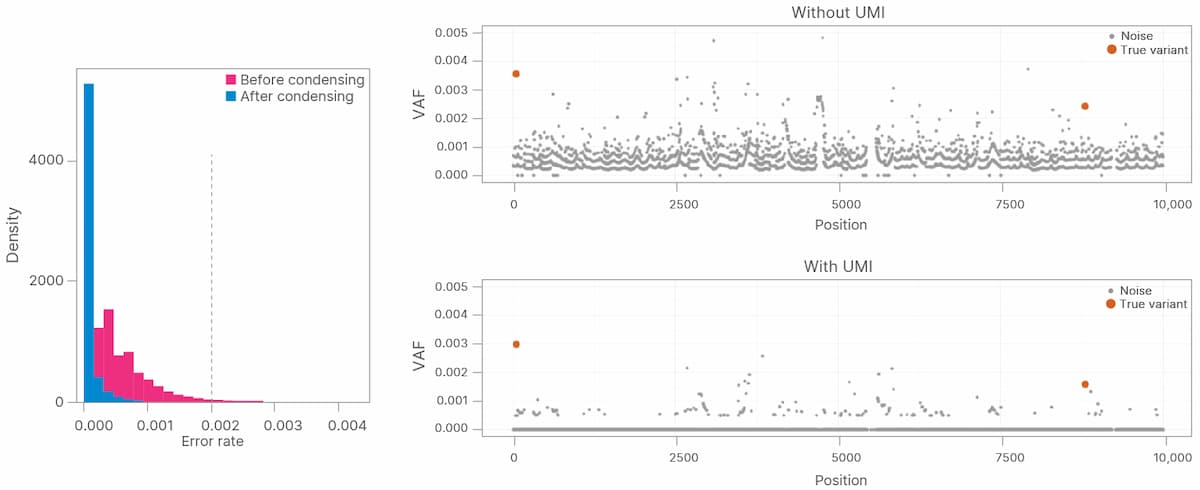
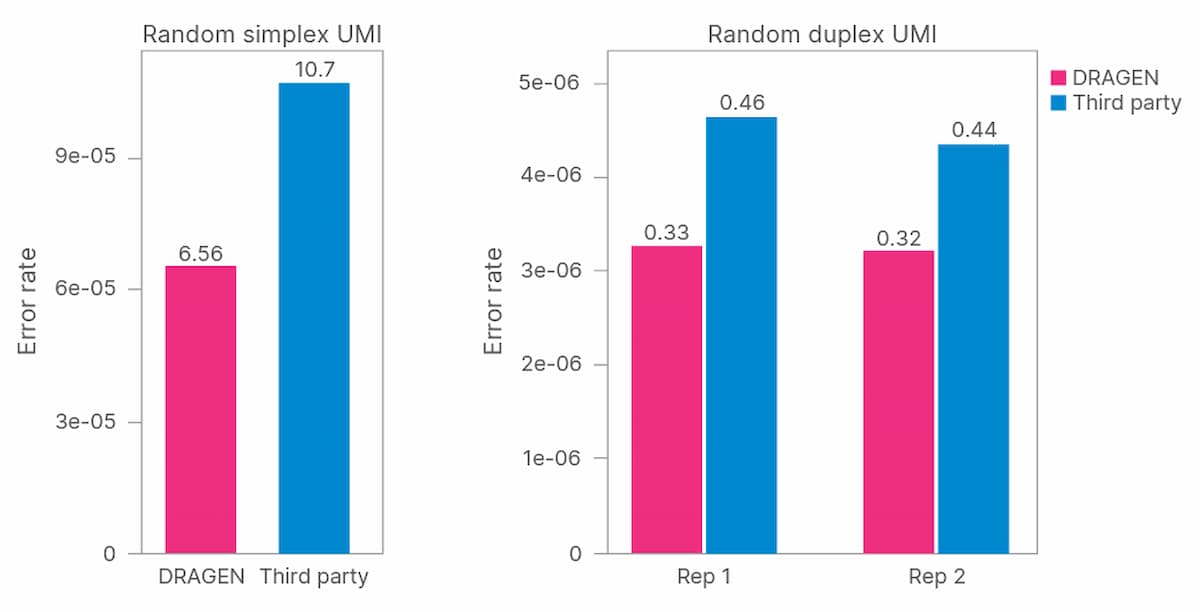
UMI barcodes were used to collapse duplicate reads (sequencing reads with the same UMI) into a single consensus sequence in which PCR duplicates and sequencing errors have been removed, while preserving low- frequency variants (Figure 1). Collapsed sequences supported by reads from one strand are referred to as simplex sequences, while those supported by reads from both the forward and reverse strand are referred to as duplex sequences. Duplex sequences have a further reduction in error rate (false positives/false negatives (FP+FN)) due to the low probability of having identical errors on both strands.
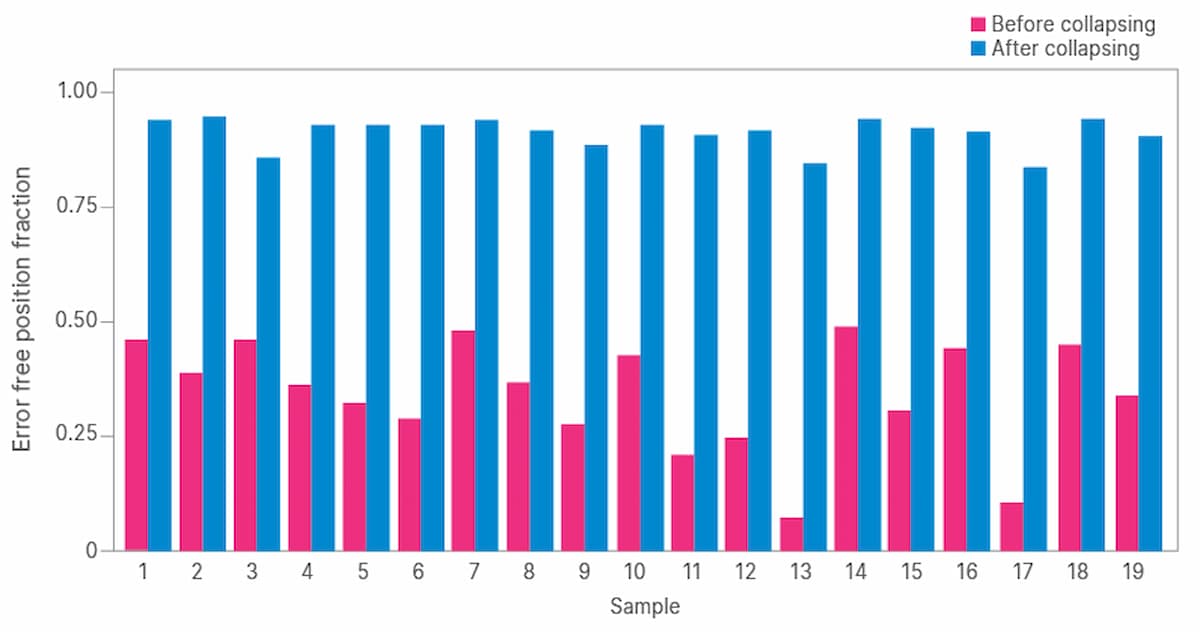
To evaluate performance of the DRAGEN platform for error correction with UMIs, two metrics were determined. The error free position fraction refers to the percentage of base calls in all sequencing reads at each genomic position that match the reference sequence. The mean error rate is similar but is averaged across all genomic positions.
Results
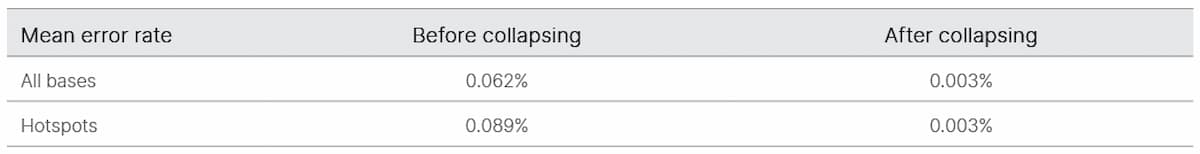
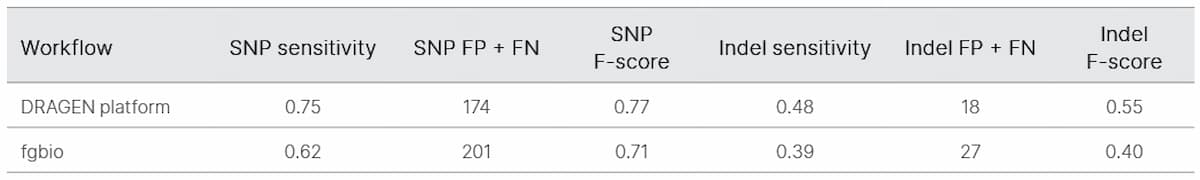
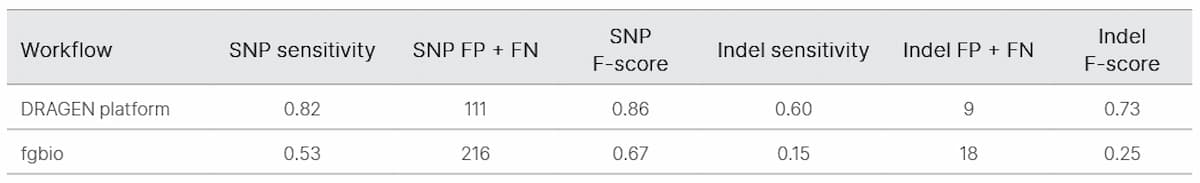
We began by evaluating TruSight Oncology UMI reagents for noise reduction in ctDNA sequencing. We determined the error-free position fraction before and after UMI collapsing across 19 samples (Figure 2). The mean percent error-free position was 36.3% before collapsing and increased to 92.9% after collapsing (Table 1).

Summary
NGS-based analysis of ctDNA by liquid biopsy can enable various oncology applications to assess tumor characteristics and better understand cancer development and progression. However, the rarity of ctDNA presents a challenge to discern noise and errors from true sequence variants. In this article we are showing how UMIs combined with the DRAGEN platform can significantly reduce errors, but there are other strategies we are using to increase accuracy such as systematic noise filtering, the artifact reduction caused by aging variants. Together, these methods and enable accurate somatic variant calling in ctDNA.
References
- 1. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies Sci Transl Med. 2014;6(224):224ra24.