Summary
- Biallelic inactivation of the CYP21A2 gene leads to autosomal recessive congenital adrenal hyperplasia (CAH), a serious and potentially life-threatening condition.
- CYP21A2 lies within a large multigene segmental duplication on chromosome six, where extremely close sequence homology has led to a high level of genomic instability across human populations.
- This homology and population complexity both cause significant interference with standard DNA sequencing methodologies.
- We have developed and integrated into DRAGEN a targeted caller with high sensitivity and specificity for small variants and genomic rearrangements in this region using whole-genome sequencing (WGS).
- This software will promote research into CAH, improving understanding of the role variants in CYP21A2 play in human health
Introduction
CYP21A2 in RCCX: A 30,000-base segmental duplication in the MHC Class III region
CYP21A2 encodes 21-hydroxlyase, a cytochrome P450 enzyme that aids in adrenal regulation of the cortisol and aldosterone hormones.1,2 These hormones play a number of roles, including in regulating salt retention in the kidneys. Inactivation of CYP21A2 is responsible for 95% of 21-hydroxlyase CAH cases,3 which can take one of three forms:
- Salt-wasting CAH is the most severe, in which complete deficiency of CYP21A2 leads to very low levels of aldosterone synthesis and thus decreased sodium retention. Symptoms can be very severe, including dehydration, diarrhea, vomiting, and adrenal crisis, and can lead to death.4 Low cortisol levels also play a developmental role and can lead to virilization.5
- Simple virilizing CAH is a more moderate form, caused by decreased CYP21A2 activity without complete gene deficiency. This form generally avoids the most severe and life-threatening symptoms, but still typically presents virilization and developmental challenges.6
- Non-classic CAH can also occur, with similar symptoms to simple virilizing CAH. Non-classic CAH is characterized by higher aldosterone and cortisol hormone levels, resulting in milder symptom severity.7 Due to the lesser phenotypic impact, non-classic CAH is more difficult to diagnose.
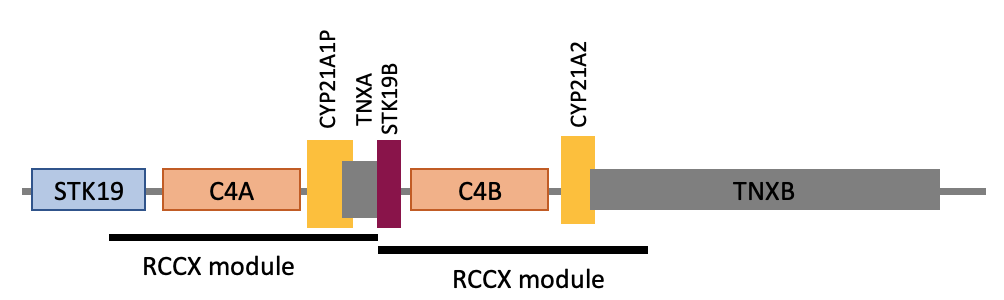
Most genomes contain two copies of the 30kb RCCX region, shown with a black bar under gene labels. CYP21A2, about 3kb in length, lies within the second copy of RCCX. Other genes with human health implications, C4A, C4B, and TNXB, are also each fully or partially inside the repeat region.
CYP21A2 lies within a 30 kilobase segmental duplication, in the major histocompatibility complex (MHC) class III region.8 The repeat is commonly referred to as RCCX and contains part or all of four genes: STK19, C4A/C4B, CYP21A2, and TNXB.9 The RCCX repeat canonically exists as two modules with nearly identical sequences (Figure 1). The first module contains the end of the STK19 gene, an active C4A gene, and two inactive pseudogenes: CYP21A1P and TNXA. The second module contains C4B, CYP21A2, and the end of TNXB, all active genes with important roles in human health.10–12
The high sequence homology of the RCCX region drives a high rate of non-allelic homologous recombination.9,13 These recombination events may occur at any point within the repeat (Figure 2). If the breakpoints of a recombination event lie within the regions of CYP21A2, a chimeric gene fusion is created with part of the sequence of the pseudogene and part of the sequence of the gene. Despite sequence similarity between gene and pseudogene of about 98%,14 these chimeric fusion genes may be partially or entirely inactivated by the introduction of a few small variants from the pseudogene into the gene. These may be considered partial gene conversions. CYP21A2 is also subject to more canonical gene conversion variants of partial gene sequences, perhaps due to template switching during break repair in synthesis.15
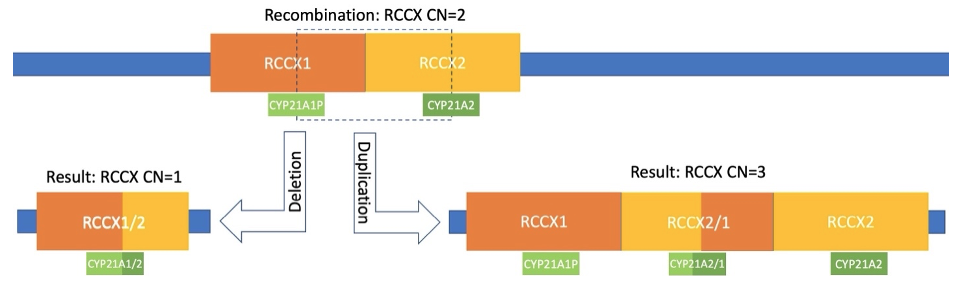
Homologous recombination between copies of the RCCX module may have variable boundaries within the repeat, resulting in either a deletion or duplication of the region. If the breakpoints of that recombination occur within the CYP21A2 gene, a chimeric gene fusion is created with part of the gene and part of the nearly identical pseudogene CYP21A1P.
If the recombination breakpoints for a deletion occur outside the gene, it may be entirely deleted from the resulting chimeric RCCX module, leaving only CYP21A1P. This heterozygous CYP21A2 deletion creates a carrier status and will result in phenotypic impacts if later co-inherited with another deficient allele.
Other small variants (single-nucleotide and insertion/deletion events) can also lead to decreased CYP21A2 activity.16 These variants occur in regions of the gene where the sequence is identical to the pseudogene, making variant detection extremely challenging. This is because reads sequenced from either the gene or pseudogene lack identifying markers, meaning they are randomly assigned in alignment and may be placed in the wrong copy of the repeat. The result is weak and ambiguous evidence for a variant in either location, which can mean missed or low-confidence variant calls. Gene conversion variants may be even more difficult to detect, as these reads will contain the allele from the alternate RCCX module at the gene conversion site and may be preferentially mapped to the wrong copy.
This combination of factors has meant that despite the impact of variant discovery in CYP21A2 for human health research, it has previously been a very difficult or impossible task with WGS data. The DRAGEN CYP21A2 caller overcomes challenges of sequence homology to discover all three types of variation here described: small variants, gene conversions, and recombination-derived whole-gene deletions.
Method
The DRAGEN CYP21A2 targeted caller identifies the number of copies of the total RCCX region, reports any recombinant CYP21A1P-CYP21A2 gene fusions, and detects 33 small variants in either the gene or the pseudogene. These variant calls include all CYP21A2 variants with multiple submitters that have been annotated in ClinVar17 as pathogenic or likely pathogenic.
Total RCCX copy number. The DRAGEN CYP21A2 caller counts the number of copies of the total RCCX region by counting reads belonging to both copies of the segmental duplication. In most cases, reads cannot be unambiguously mapped to either copy of the repeat due to high sequence homology, but counting all reads placed in either copy provides a highly accurate measure of the summed copy number of both regions. The region used for copy number calling covers most, but not all, of the RCCX region. The region begins after a polymorphic 6.4 kb HERV-K retrotransposon in introns of both C4A and C4B and extends 20 kb downstream to a 120 bp deletion in TNXA, including the entirety of the CYP21A2 gene. The copy number calling subregion of RCCX is therefore large enough to reach any nonallelic homologous recombination events (which, affecting a whole copy of RCCX, are 30 kb in length). Read coverage is corrected for GC content by normalization against a panel of 3000 preselected 2 kb genomic sites with highly consistent diploid copy number. This normalized copy number is an accurate estimate of the total copy number of the RCCX segmental duplication.
Recombinant variant detection. The caller uses a panel of 18 sites across CYP21A2 to detect gene fusions between CYP21A1P and active CYP21A2. These are sites where the sequence of the gene and the pseudogene differ. Fifteen protein-altering gene conversion variants are included, as well as seven non-protein-altering sites.
Identifying recombinant variants requires detecting the haplotypes that occur within the genome. To do this, the caller collects reads that span the set of 18 differentiating site variants. Reads that span multiple sites are used to build connected haplotypes across the entire region (Figure 3).
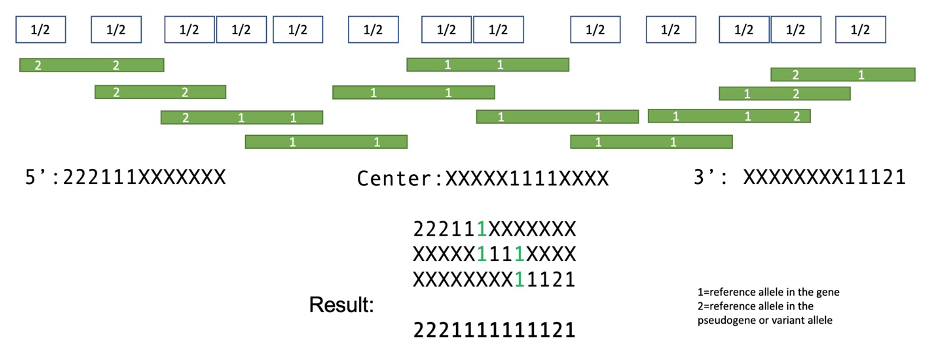
Reads containing sites that differ between gene and pseudogene are collected and assembled into partial haplotypes from the 5’ end, center, and 3’ end of the gene. Partial haplotypes are then assembled into final complete haplotypes that span the full gene region. Transitions within the resulting haplotypes, from gene-allele to pseudogene-allele sequences, may indicate either full chimeric gene fusions or smaller gene conversion events.
Targeted small variant detection. Other deleterious variations are detected for a set of 33 known sites, where the gene and pseudogene sequences are identical. Reads aligning to the gene or pseudogene for each of these sites are collected. The number of reads containing the reference allele, and any supporting the deleterious alternate allele, are counted and reported. The reads are then used to provide evidence for the presence or absence of the pathogenic allele in either the gene or the pseudogene.
Results
We tested the DRAGEN CYP21A2 caller on a large selection of genomes, including CAH cases, carriers, and healthy genomes from the 1000 Genomes Project (1KG).18
CAH cases from the Radboud UMC (N = 16, cases): Collaborators at Radboud University Medical Center shared WGS data for 16 CAH cases with validation from Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA). In each of these case genomes, the DRAGEN CYP21A2 caller was able to detect the total RCCX copy number and the pathogenic variants, including small variants, full gene deletions, and inactivating gene conversions (Table 1).
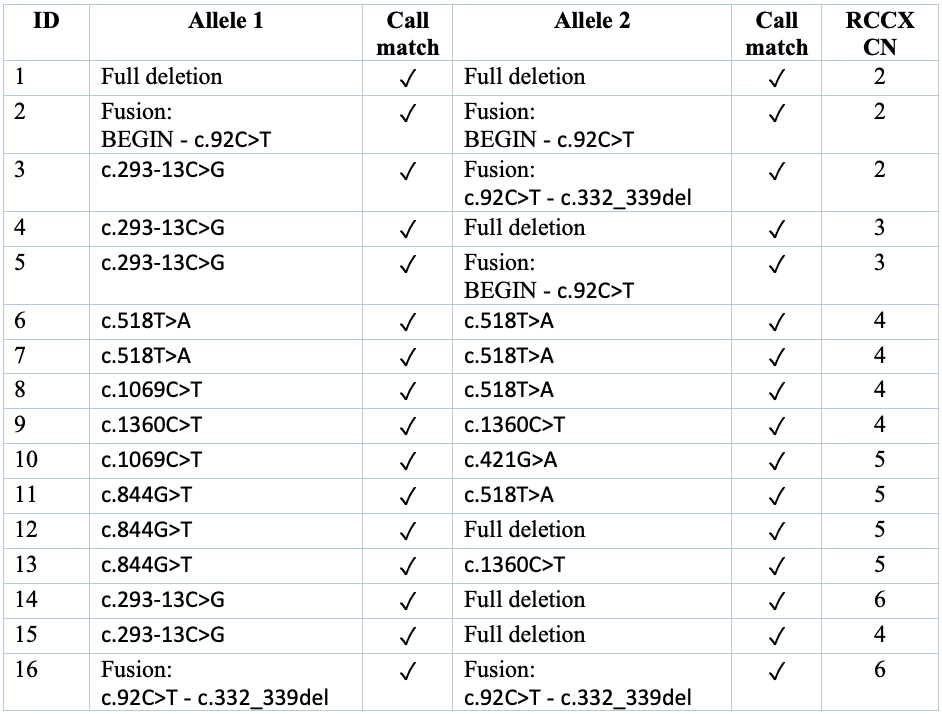
In each genome, DRAGEN reported the causal alleles and total RCCX copy number. DRAGEN calls matched MLPA/Sanger results in each case. All variant IDs are respective to the NM_000500.9 transcript.
Cell lines from the Coriell Institute (N = 4, cases and carriers): We also tested the DRAGEN CYP21A2 caller on four sequenced cell lines, with MLPA or long-range PCR confirmation of CYP21A2 variants, from the Coriell Institute for Medical Research. These included a trio in which the proband, NA14734, was affected by the severe salt-wasting form of CAH. This was caused by full deletion of two copies of the RCCX segmental duplication and a complete loss of CYP21A2, as evidenced by MLPA validation. MLPA also revealed that both parents were carriers of CYP21A2 deletions, clarifying the inheritance of the deleterious genotype in the proband.
DRAGEN identified each of these genotypes in the trio, reporting the haplotypes generated by deletions of the RCCX module and the total RCCX copy number in each family member. The detailed information obtained from the DRAGEN-reported haplotypes also provides insight into the inherited alleles of the CYP21A1P pseudogene (Figure 4). Each parent can be identified as a possible CAH carrier due to decreased RCCX copy number. The proband, lacking any copies of the active gene, is identified as a likely CAH case.
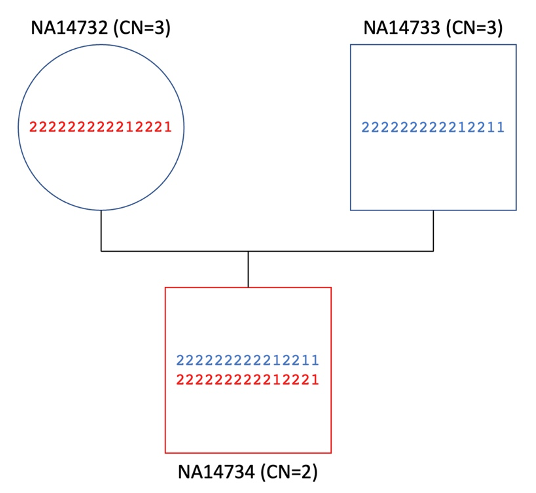
Each haplotype is simplified to a series of 1 or 2 identifiers, indicating the gene (1) or pseudogene (2) case at each differentiating site. The CAH-affected proband NA14734 contains copies of the RCCX segmental duplication with the inactive pseudogene CYP21A1P case at most sites, and no copies of the wildtype CYP21A2 gene. DRAGEN results identify the most likely parental origins of the two RCCX copies in the proband (with inheritance color-coded). Copy number calls of 3 in each parent also indicate risk of wildtype gene deletions.
The fourth CAH cell line acquired from the Coriell Institute (NA12217) was also a CAH case, although affected by the more moderate simple virilizing form of the disorder. In this genome, MLPA and long-range PCR validation identified a single deletion of one copy of RCCX and an exonic single-nucleotide variant, NM_000500.9:c.518T>A, with known CAH risk. DRAGEN identified the NM_000500.9:c.518T>A variant in one allele, reported the total RCCX copy number of four, and also identified a chimeric pseudogene-gene fusion likely derived from a recombination-mediated deletion. This deletion event, in tandem with the total RCCX copy number of four, indicates that this genome represents the outcome of both deletion and duplication in the RCCX module. The chimeric fusion haplotype structure can be represented as “222222211111111111”, where “1” indicates the target gene allele and “2” indicates the pseudogene allele. This shows a clear delineation between consistent pseudogene alleles at the first seven differentiating sites, then conversion to consistent gene alleles at the final eleven sites, a refined representation of the fusion gene structure and deletion breakpoints.
1KG genomes with orthogonal RCCX copy number calls (N = 204). We compared the RCCX total copy number calling results from the DRAGEN CYP21A2 caller against RCCX copy number calls from the orthogonal Bionano Genomics optical mapping technology in 204 genomes from the 1KG cohort (Figure 5). While optical mapping lacks the resolution to identify gene fusions or small variation, these call comparisons indicate the overall copy number calling accuracy of the DRAGEN CYP21A2 caller. In 201 of 204 genomes, the copy number calls agreed, while in three genomes there was a disagreement of one RCCX copy. This concordance demonstrates the high accuracy of the DRAGEN CYP21A2 caller in recovering the correct copy number of the RCCX region.
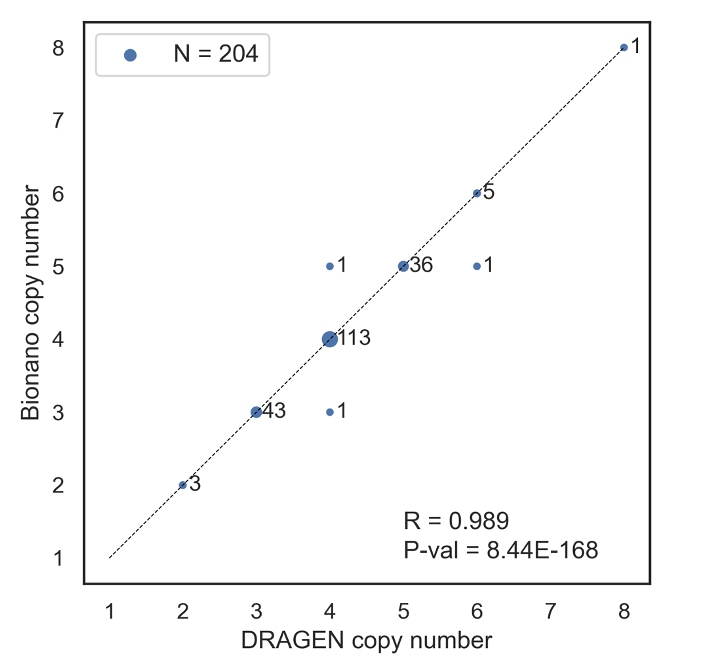
Pearson’s correlation coefficient and P-value annotated at bottom right.
Targeted small variants in 1KG: We ran the DRAGEN CYP21A2 caller on 3195 samples from the 1000 Genomes Project cohort and reviewed results for the 33 small variants that DRAGEN targets for CYP21A2. Eleven out of 3195 (0.3%) contained strong evidence for a targeted variant (at least two supporting reads, from either the gene or pseudogene). While these variant calls are highly confident, they cannot be assigned to the gene or pseudogene without confirmatory testing.
Availability
The CYP21A2 caller will be available in the 4.2 release of DRAGEN. Please contact ffg-info@illumina.com to request early access to the software.
Acknowledgments
We thank Gaby Schobers at Radboud University Medical Center.
References
- Pignatelli, D. et al. The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front Endocrinol (Lausanne) 10, 432 (2019).
- Torres, N. et al. Phenotype and genotype correlation of the microconversion from the CYP21A1P to the CYP21A2 gene in congenital adrenal hyperplasia. Braz J Med Biol Res 36, 1311–1318 (2003).
- Huynh, T. et al. The Clinical and Biochemical Spectrum of Congenital Adrenal Hyperplasia Secondary to 21-Hydroxylase Deficiency. Clin Biochem Rev 30, 75 (2009).
- Khanal, D., Mandal, D., Phuyal, R. & Adhikari, U. Congenital Adrenal Hyperplasia with Salt Wasting Crisis: A Case Report. JNMA J Nepal Med Assoc 58, 56 (2020).
- Kovács, J. et al. Lessons From 30 Years of Clinical Diagnosis and Treatment of Congenital Adrenal Hyperplasia in Five Middle European Countries. J Clin Endocrinol Metab 86, 2958–2964 (2001).
- Singh, R., Agarwal, M. & Sinha, S. Challenges in the Diagnosis of Simple-Virilizing Congenital Adrenal Hyperplasia: A Case Report. Cureus 14, (2022).
- Bidet, M. et al. Clinical and Molecular Characterization of a Cohort of 161 Unrelated Women with Nonclassical Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency and 330 Family Members. J Clin Endocrinol Metab 94, 1570–1578 (2009).
- Schubert, T. et al. CYP21A2 Gene Expression in a Humanized 21-Hydroxylase Mouse Model Does Not Affect Adrenocortical Morphology and Function. J Endocr Soc 6, (2022).
- Carrozza, C., Foca, L., de Paolis, E. & Concolino, P. Genes and Pseudogenes: Complexity of the RCCX Locus and Disease. Front Endocrinol (Lausanne) 12, 941 (2021).
- Pereira, K. M. C. et al. Impact of C4, C4A and C4B gene copy number variation in the susceptibility, phenotype and progression of systemic lupus erythematosus. Adv Rheumatol 59, 36 (2019).
- Baş, F. et al. CYP21A2 gene mutations in congenital adrenal hyperplasia: genotype-phenotype correlation in Turkish children. J Clin Res Pediatr Endocrinol 1, 116–128 (2009).
- Merke, D. P. et al. Tenascin-X Haploinsufficiency Associated with Ehlers-Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab 98, E379–E387 (2013).
- Carvalho, C. M. B. & Lupski, J. R. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17, 224 (2016).
- Parajes, S., Quinteiro, C., Domínguez, F. & Loidi, L. High Frequency of Copy Number Variations and Sequence Variants at CYP21A2 Locus: Implication for the Genetic Diagnosis of 21-Hydroxylase Deficiency. PLoS One 3, e2138 (2008).
- Chen, J. M., Cooper, D. N., Chuzhanova, N., Férec, C. & Patrinos, G. P. Gene conversion: mechanisms, evolution and human disease. Nature Reviews Genetics 2007 8:10 8, 762–775 (2007).
- Krone, N., Riepe, F. G., Grötzinger, J., Partsch, C. J. & Sippell, W. G. Functional characterization of two novel point mutations in the CYP21 gene causing simple virilizing forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 90, 445–454 (2005).
- Landrum, M. J. et al. ClinVar: improvements to accessing data. Nucleic Acids Res 48, D835–D844 (2020).
- Byrska-Bishop, M. et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 2021.02.06.430068 (2021) doi:10.1101/2021.02.06.430068.