Genomic Surveillance Methods

识别和追踪传染性疾病威胁
使用新一代测序(NGS)进行基因组学监测可追踪传染性疾病传播,鉴定冠状病毒新毒株和其他新出现的病原体。利用几乎完整的病原体基因组测序数据,我们可以实施有效的传染性疾病监测策略,帮助预防进一步的传播和感染。
利用NGS进行传染性疾病监测可以:
- 跟踪传染性疾病在全球的传播途径
- 确定确定冠状病毒等病原体在传播过程中的突变速率
- 鉴定病原体中的新型和已知突变,如冠状病毒、甲型流感和乙型流感等
- 研究传染性疾病治疗耐受性
- 研究疫苗逃逸机制
基因组学监测有助于公共卫生人员追踪疫情路径途径,以及病原体当下的变化是否会影响诊断效果或治疗效果。
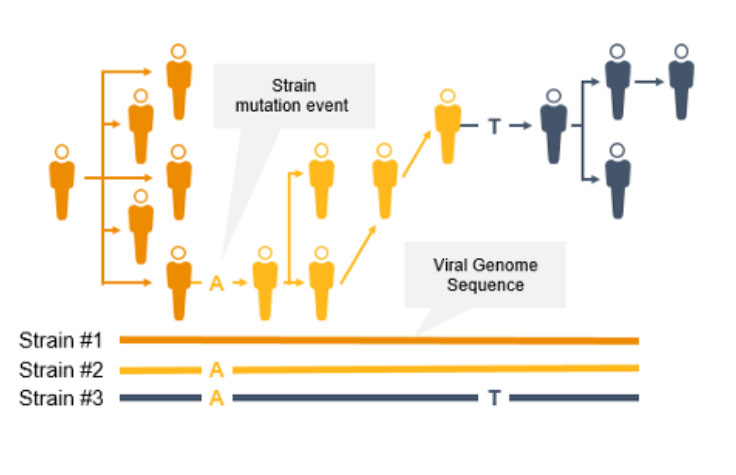
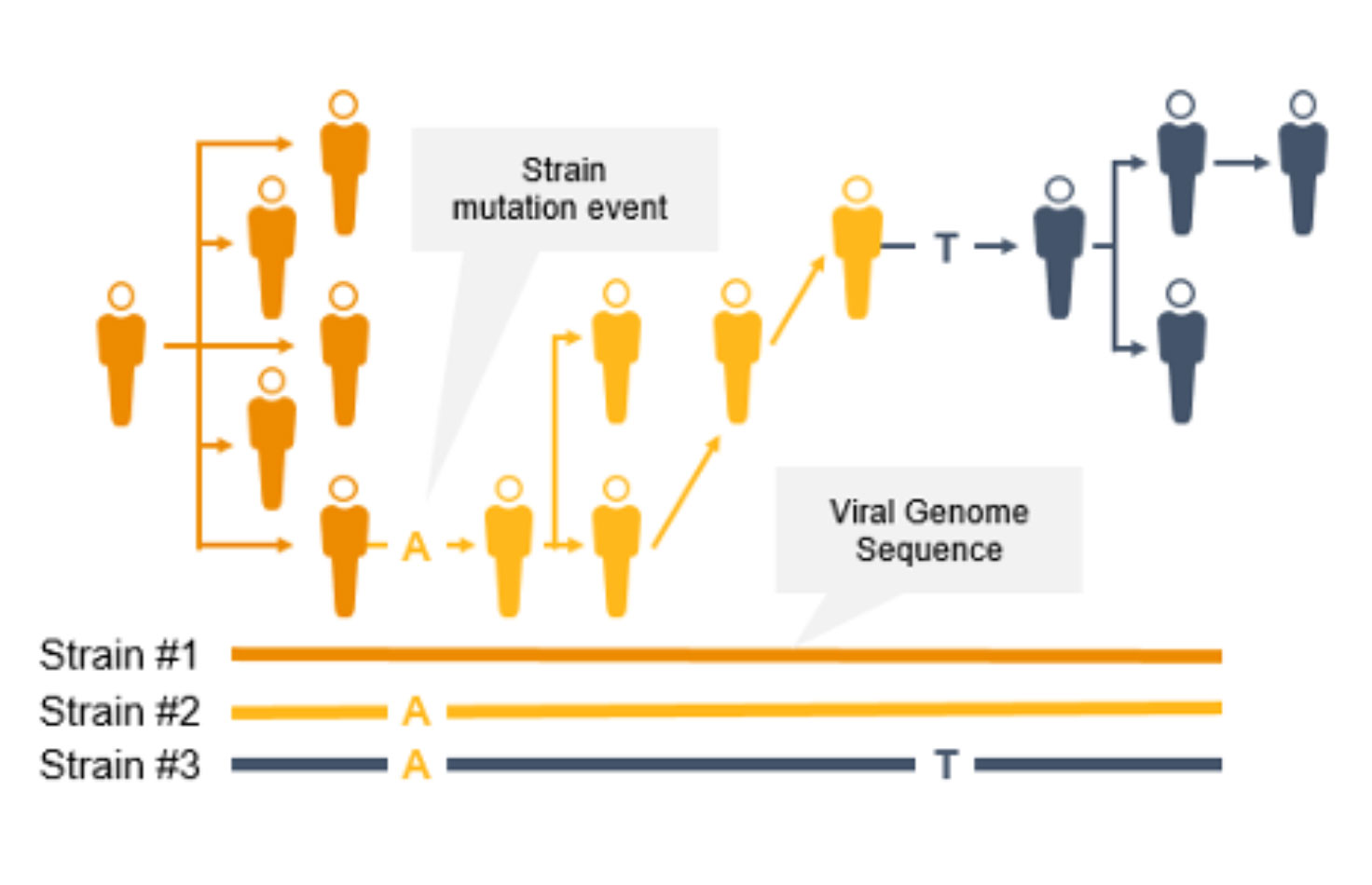
监测和鉴定冠状病毒突变
随着冠状病毒的变异,新的变体不断进化,例如阿尔法(B.1.1.7,英国)、贝塔(B.1.351,南非)、伽玛(P1,巴西)、德尔塔(B.1.617.2,印度)和奥密克戎(B.1.1.529,南非)变种。监测和监控极为重要,因为突变可能导致更强的传播性或传染性。这些冠状病毒突变可能会降低疫苗的有效性/保护性,甚至逃避检测诊断。
NGS是一项用于COVID-19等传染病基因组监测的重要技术。与PCR技术不同,NGS不仅能追踪冠状病毒突变毒株的传播,还能鉴定新型冠状病毒突变。使用NGS,科学家可以检测低频次要变异和多种多态性以及新变异。
NGS全基因组菌株分型可以快速识别并鉴定突变,从而防止进一步传播。菌株水平的追踪可支持爆发聚集地和传播途径的识别。
相反,PCR旨在检测病原体基因组特定区域,不会在这些快速进化的病原体基因组中识别出新的突变。此外,如果引物或探针结合区域发生突变,PCR的性能可能会受到影响。
疫情无处不在
猴痘病毒(MPXV)是一种大型双链DNA病毒,于1958年首次发现。历史上,猴痘病毒曾在中非和西非给人们带来了零星的疫情,死亡率在1%至10%之间[1]。2022年春季,在欧洲、北美和东南亚的非流行国家观察到疫情。这些国家正在开展公共卫生监测工作,从而防止猴痘病毒的进一步传播。
NGS是一项用于猴痘等传染病基因组监测的重要技术。NGS不仅可以追踪突变菌株的传播,还可以识别新的突变。因此,科学家现在可以采用基于NGS的技术以高通量规格快速、准确地检测低频次要变异、多种多态性以及新型突变。这些因素对于协助流行病学家识别和表征爆发聚集地、传播途径和突变方面是至关重要的,可防止进一步传播。
因美纳很自豪能够在COVID大流行期间支持NGS功能和公共卫生功能的全球扩张。这些资源现在可用于监测猴痘病毒、埃博拉病毒、结核分枝杆菌等病原体。
比较NGS病原体监测方法
| 检测需求 | 扩增子 | 靶向富集 | 鸟枪法宏基因组学测序 |
|---|---|---|---|
| 速度与周转时间 | |||
| 可扩展且经济高效 | |||
| 识别新病原体 | |||
| 追踪传播 | |||
| 检测突变 | |||
| 鉴定合并感染和复杂疾病 | |||
| 检测抗菌药物耐药性 |
充分满足实验室的检测需求
部分满足实验室的检测需求
有关传染病监测测序方法的更多信息
扩增子测序
检测并全面表征已知病毒。通过扩增子方法进行全基因组测序对于基因组较小的已知病毒来说是理想的选择。该方法会使用PCR扩增子的超深度测序来分析目标基因组区域。
靶向富集测序
检测和表征冠状病毒、流感病毒和其他病原生物,以及相关的抗菌药物耐药性等位基因。这些信息可以帮助公共卫生人员监测疫情并优化感染控制策略。该方法通过与特定靶点探针的杂交捕获目标基因组区域。
鸟枪法宏基因组学测序
通过对给定样本中的所有生物体进行全面测序,鉴定出像猴痘病毒这样的新兴病原体。这种NGS方法有助于加速疫情调查,并支持新实验室检测方法的开发。
FAQs
特色产品

COVIDSeq家族
COVIDSeq检测和COVIDSeq测试是基于引物扩增的新一代测序(NGS)方法,旨在帮助公共卫生实验室鉴定SARS-CoV-2的新型毒株。

iSeq 100
iSeq 100系统利用了快速、价优的互补金属氧化物半导体(CMOS)技术,以及准确的Illumina边合成边测序(SBS)化学技术。

NextSeq 2000
能助您探索一系列现有及新兴应用,突破效率,突破极限的开创性台式测序仪。
预防下一次大流行
据估计,多达75%的新型或新出现的传染性疾病具有人畜共患疾病传染源1,2。我们现在知道,人畜共患疾病宿主在病原体传播中起着重要作用。有了NGS,人们能够筛查蝙蝠等宿主动物,从而预测和预防病毒病原体的爆发。
利用基于NGS的靶向富集或宏基因组学进行传染性疾病监测可帮助我们了解跨物种传播、人畜共患传染性疾病出现方式、传播和对常见疗法产生耐受性,并使我们能够更好地控制、治疗和预防疾病爆发。
靶向富集测序可放大至监测人畜共患疾病病原体,而宏基因组学基因组监测支持对多种病原体进行无偏差、无培养的检测和鉴定。宏基因组学还能帮助我们了解微生物组和病原体相互作用之间的关系,这对于控制传染性疾病措施的制定十分重要。
了解详情:
靶向富集宏基因组学测序
更多传染性疾病监测应用
冠状病毒监测
NGS可以对新的冠状病毒毒株进行无偏差鉴定。因美纳提供SARS-CoV-2冠状病毒突变的快速检测,可满足高效测序的需求。
食源性疾病监测
微生物全基因组测序可评估从食品传播到人类的病原体的全部遗传变异信息。我们能识别的变异越多,找到感染源和传播途径的可能性越高。
病毒监测
靶向富集使用杂交捕获目标基因组区域。此方法可提供检测病毒所需的高灵敏度,并提供病毒传播和进化的相关信息。
呼吸道感染监测
这些panel可提供呼吸道病原体和呼吸道病毒检测和鉴定的靶向富集测序解决方案,包括COVID-19和流感。
人畜共患疾病宿主监测
鸟枪法宏基因组学测序能够对人畜共患疾病样本中来自完整微生物群落的DNA进行测序,包括新型物种和已知物种。这些测序read可用于了解AMR基因的出现、进化和传播及物种分布。
医疗相关感染监测
基于NGS的细菌基因组测序结合用户友好的bioMérieux软件,可进行全面的分离株区分和鉴定。
Frequently Purchased Together
特色基因组监测文献
猴痘病毒多重PCR扩增子测序(PrimalSeq)V.2。
2022年猴痘病毒多国爆发的系统基因组学特征和微进化迹象
污水基因组测序检测区域性流行SARS-CoV-2变异株
为了及早检出新出现的菌株需要进行大规模基因组监测
通过监测和基于探针捕获的NGS发现蝙蝠冠状病毒
参考文献
- Woolhouse ME, Gowtage-Sequeria S. Host range and emerging and reemerging pathogens. Emerg Infect Dis. 2005;11:1842–7. 10.3201/eid1112.050997
- Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990–3.