本文是与Severine Catreux(Illumina DRAGEN团队)、James Emery(Broad研究所GATK团队)和GATK支持团队的成员合作完成的。
去年9月,我们宣布了一项与Illumina DRAGEN团队共同开发分析方法和流程的新合作项目。如果您当时没有看到这条新闻,可以查看当时的博客文章,了解合作的背景和目标。目前,我们正在合作开发用于NGS数据做变异检出的统一DRAGEN-GATK流程,它将同时以免费开源版本(通过Broad)和许可证硬件加速版本(通过Illumina)提供。
在这里,我想介绍一下DRAGEN团队开发的部分算法改进,我们正在将其迁移到开源GATK包。

DRAGEN以快速著称
DRAGEN在后台使用了一种称为FPGA(现场可编程门阵列技术)的硬件,可为其基于GATK的生殖系短变异发现流程提供极快的加速。我们在这里暂不讨论过多细节,仅从时间上看,从未经比对的原始FASTQ文件开始并生成GVCF和/或经过过滤的VCF,单个全基因组样本的端到端平均分析时间可从23小时以上缩短到约36分钟。不用说,那很酷。但要说明一点,我们的合作并不仅仅只为了实现GATK作为一个软件工具的运算加速。如果您有这种数量级的加速需求,您可能需要了解Illumina的商业化DRAGEN产品,它包含硬件加速版本。这项合作明确可带来的益处是,在开源纯软件版本(标记为DRAGEN-GATK)中实现一些具体的准确性提升。当然,这里也有一个好消息:两个版本(纯软件版及硬件加速版)的输出功能相当,并兼容跨数据集分析。
DRAGEN因准确而卓越
我承认,在得知有人试图将我们的工具在速度上进行提升的时候,我也总是担心这是否会牺牲工具的准确性。我为什么担心?首先,重新实现复杂算法的难度很大。在不同编程语言间转换时可能会丢失一些细节,即使经验丰富的开发人员也可能错误地认为某些代码低效、不重要而将其去除,却没有意识到这些代码实际上在处理复杂极端情况时具有重要的作用。与之相关的验证非常困难;不能仅凭一个样本的一个高质量测序运行就宣布一切正常。验证上必须要运行包含多种异常和潜在质量问题的各种不同的数据集,并确认仍能处理所有怪异的临界情况。
DRAGEN团队的优势之一在于他们一开始就理解这一点,他们使用了许多数据集,覆盖了不同患者、文库制备试剂盒、测序仪、读长和覆盖深度,以确保在多种测序条件下其方法均具有稳健性。此外,该团队的目标不仅仅是达到原GATK工具的准确性,并且也采取了措施去尝试提升原有的准确性。而这也正是故事的精彩之处。
在分析了与真值集进行对比后的的大量假阴性和假阳性结果的数据集后,DRAGEN团队发现了两个可用方法:他们决定改善插入缺失误差模型以应对PCR引起的STR区域的片段滑移,并通过累积误差模型改进基因分型算法。下面我们简单了解一下这两方面。
新插入缺失误差模型解决PCR引起的STR区域片段滑移
变异检出中的主要困难在于区分不同类型的误差(由于污染、文库制备杂峰、测序杂峰、错误比对造成)以及真实变异。插入缺失可靠检出的难度尤其巨大,因为在短串联重复序列(STR)存在时,插入缺失错误更可能发生,错误率可能取决于STR的周期和长度。和许多其他变异检出软件一样,HaplotypeCaller使用隐马尔科夫模型(HMM),使用预先确定的非样本特异性的函数对误差的统计行为建模,作为概率计算的一部分。
DRAGEN团队发现,通过在变异检出前增加样本特异性的逻辑并详细考虑STR周期和长度,可以对插入缺失错误过程进行更准确的建模。样本特异性逻辑是在BAM输入上运行的自动校准步骤,用于确定插入缺失错误和插入缺失变异的先验概率。测试发现灵敏度和特异性方面均展示出了更好的检测性能:成功发现此前遗漏的检出,并去除了此前检出的假阳性。
检测相关累积误差提高基因分型准确性
与大部分变异检出软件类似,HaplotypeCaller假设测序和/或比对错误在read之间是独立的。根据这一假设,不太可能出现多个相同错误发生在同一基因位点的情况。但是,观察到突发错误远比独立假设的预测常见,这些突发错误可造成大量假阳性。
减少这一问题发生的经典方法是在基因分型程序上游设置检查点,过滤不能满足某些质量阈值的read,例如MAPQ过低的情况。但这并非解决问题的最优方法,因为它从变异检出软件隐藏了有价值的证据。这可能导致变异检出软件遗漏真正的变异,并且难以抑制许多假阳性的检出。
DRAGEN团队开发了一种不同的方法解决这一问题,它包含两个主要部分:1)增强基因分型程序,使其能够鉴定相关累积误差并引入新的基因分型假设,以及2)在基因分型程序增强的基础上,打开上游的检查点,接受此前拒绝的read进入基因分型过程。同样,团队发现了此前遗漏的变异并去除了此前检出的假阳性。
该方法带来了一个有趣的结果,DRAGEN团队发现,这些对基因分型的改进允许他们简化变异检出后仍要进行的过滤,其原因部分在于基因分型程序中引入相关的噪声误差模型提高了QUAL评分的准确性,使其进入真值范围。因此,DRAGEN具有非常简单的仅基于QUAL评分的硬过滤规则。
GATK团队评估确认准确性提升
我们的团队与DRAGEN团队使用的评估方法略有不同,因此,我们希望在自己的测试框架下进行评估,以确认这些提升成果的表现。我们使用Illumina借出的服务器对我们最喜欢的测试样本运行了DRAGEN流程(终于体验到了这非凡的速度——快,名副其实!),概括来讲,我们的测试也在灵敏度和特异性方面得到了同样的总体提升,具体请见下图。
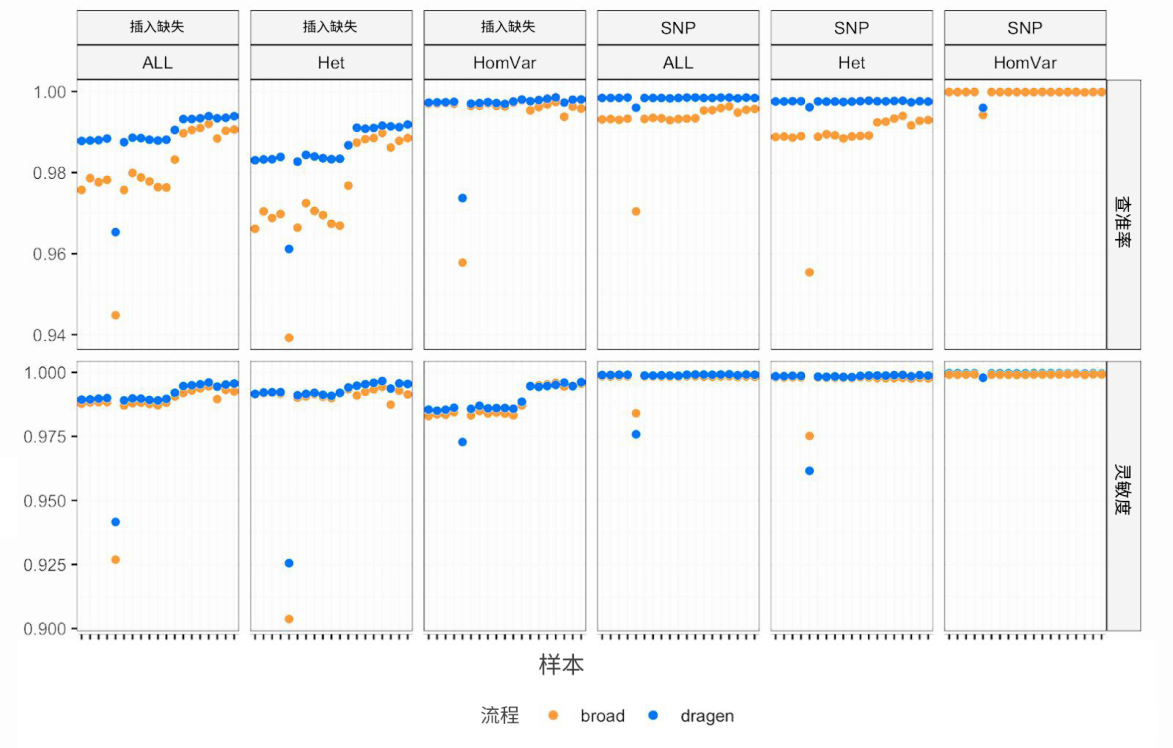
(在博客文章中将使用全尺寸图像)
简言之,图中展示了NA12878样本的19个技术平行重复(不同测序运行)与Genome In A Bottle(GIAB)真值集(高置信区域)相比的灵敏度和特异性。可以看到,DRAGEN流程(蓝色)与我们原始的带2D CNN过滤的GATK单基因组流程(橙色)相比,插入缺失和杂合子SNP检出准确性明显提升,同时,插入缺失检出灵敏度也具有相应的提升,这也是令人赞许的改善。不错的成果!
获得硬件加速的DRAGEN-GATK
根据以上结果,我们认为我们测试的DRAGEN流程(v 3.4)是我们期望达到的标准,因此,我们考虑将其作为DRAGEN-GATK生殖系短变异流程的硬件加速1.0版。换言之,如果您使用的是DRAGEN 3.4,恭喜!您一直在运行DRAGEN-GATK流程:)
我们的团队现在正努力将DRAGEN团队开发的改进内容迁移到我们的开源纯软件版本中以实现功能上的相当——这意味着您可以在我们认为不重要的误差界限内,用任何一个版本得到相同的结果。迁移工作主要涉及对HaplotypeCaller的修改,它将作为常规GATK软件包的一部分发布。完整的纯软件DRAGEN-GATK流程将作为WDL工作流程脚本在GitHub上的gatk工作流程中集中发布。
在这一工作完成后,您可将经过任一流程分析的样本合并进入下游分析,而不需要担心批次效应。特别是如今大量数据集的快速出现,这样的应对方法相当重要。不用说,没有人会纯粹为了好玩而重新处理100,000个样本。
正如您所见,我对这一合作取得的进展十分激动,因为就在开始后短短几个月的时间里,已经出现了明确的益处。接下来将有更多令人振奋的进展更新,请持续关注我们完成当前工作后的更新发布,敬请期待下一波冲击性的好消息吧!
创新的技术
在Illumina,我们的目标是应用创新技术来分析遗传变异和功能,实现仅几年前甚至还无法想象的研究。对我们而言,关键任务是带来创新、灵活且可扩展的方案,以满足我们客户的需求。作为一家重视合作互动、快速交付解决方案,和提供高质量水平的全球性公司,我们努力应对这一挑战。Illumina创新的测序和芯片技术正在推动生命科学研究、转化和消费者基因组学以及分子诊断中的进展。
仅供科研使用。不能用于体外诊断(除特殊标注)。
© 2020 Illumina, Inc. All rights reserved.